General Neurosurgery
Case Report
- Department of Neurology, Pontifical Catholic University of Sorocaba, Sorocaba, São Paulo, Brazil
- Department of Postgraduate Studies, State Public Server of São Paulo Hospital, São Paulo, Brazil
- Department of Neurosurgery, Santa Paula Hospital, São Paulo, Brazil
- Specialist Neurophysiologist-Clinical, São Paulo, Brazil
Correspondence Address:
Julia Pinheiro Martinez Serrano
Specialist Neurophysiologist-Clinical, São Paulo, Brazil
Julia Pinheiro Martinez Serrano
Specialist Neurophysiologist-Clinical, São Paulo, Brazil
DOI:10.25259/SNI-76-2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
How to cite this article: Julia Pinheiro Martinez Serrano, Maick Willen Fernandes Neves, Cassiano Marchi, Fabio Jundy Nakasone, Marcos Vinicius Calfat Maldaun, Paulo Henrique Pires de Aguiar, Wilson Scappini. Giant dumbbell C2C3 neurofibroma invading prebulbar cistern: Case report and literature review. 10-May-2019;10:77
How to cite this URL: Julia Pinheiro Martinez Serrano, Maick Willen Fernandes Neves, Cassiano Marchi, Fabio Jundy Nakasone, Marcos Vinicius Calfat Maldaun, Paulo Henrique Pires de Aguiar, Wilson Scappini. Giant dumbbell C2C3 neurofibroma invading prebulbar cistern: Case report and literature review. 10-May-2019;10:77. Available from: http://surgicalneurologyint.com/surgicalint-articles/giant-dumbbell-c2c3-neurofibroma-invading-prebulbar-cistern-case-report-and-literature-review/
Abstract
Background:Neurofibromatosis 1 (NF1) has a broad spectrum of clinical manifestations, most typically involving café-au-lait spots and skin neurofibromas. Only 2% of patients with NF1 have symptomatic spinal tumors.
Case Description:A patient with a previous diagnosis of NF1 presented with cervicalgia, dysphagia/mild dysphonia, gait alteration, and progressive hypoesthesia involving all four limbs. The magnetic resonance documented a giant dumbbell neurofibroma arising between the C2 and C3 levels which extended toward the foramen magnum, causing medullary and bulbar compression. The major challenge of surgical management was the enormous size and location this C2–C3 (5 cm × 4 cm × 5.1 cm) lesion.
Conclusions:Compression of the foramen magnum attributed to a dumbbell giant spinal neurofibroma at the C2C3 level resulting in prebulbar cisterns should be among the differential diagnostic considerations for patients presenting with tetraparesis and underlying NF1.
Keywords: Dumbell neurofibroma, Giant neurofibroma, Neurofibromatosis 1, Spinal tumors
INTRODUCTION
Type 1 neurofibromatosis (NF1) is an autosomal dominant genetic disorder caused by biallelic suppression of the NF1 tumor suppressor gene, located on chromosome 17. It occurs in 1/3,500 individuals and carries a 3%–15% mortality rate.[ 3 , 8 ] NF1 occurs in all races, and some report a small female preponderance.[ 1 , 2 , 5 ] Only 2% of patients with NF1 have symptomatic spinal tumors.
CASE REPORT
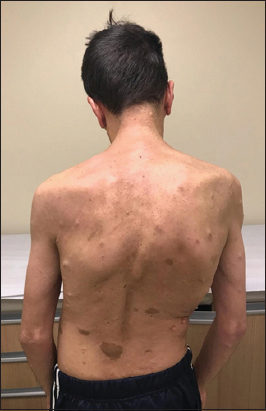
A 38-year-old male presented with a 4-year history of cervicalgia, dysphagia/mild dysphonia, gait alteration, and progressive hypoesthesia of all four extremities. The patient had a history of type I NF1 diagnosed 20 years previously. On examination, the patient exhibited cutaneous nodules/café-au-lait spots scoliosis, and a Grade 4 tetraparesis characterized by diffuse hyperreflexia with bilateral Babinski signs [ Figure 1 ].
Radiographic analysis
The cervical magnetic resonance imaging (MRI) demonstrated a solid, nodular, expansive lesion originating at the C2–C3 (5 cm × 4 cm × 5.1 cm) level with extension toward the foramen magnum. It contributed to medullary and bulbar compression [ Figure 2 ]. A secondary lesion was visualized at the C4–C5 level on the right.
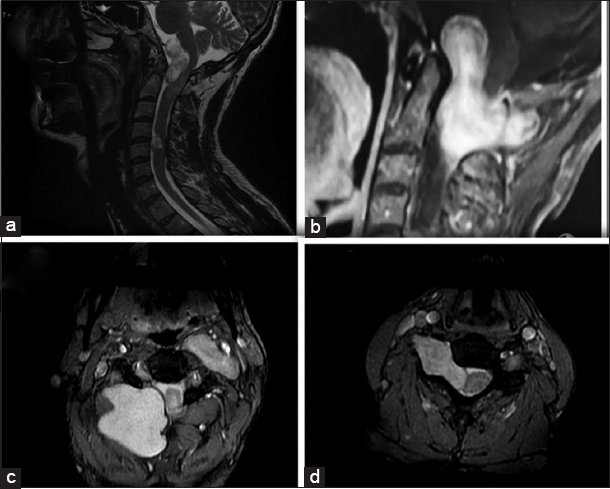
Figure 2
(a) Cervical magnetic resonance imaging – T2 with solid nodular expansive lesion originating from the level of C2–C3 (5 cm × 4 cm × 5.1 cm) with extension toward the foramen magnum, causing medullary and bulbar compression and another lesion from the level of C4–C5. (b) Dumbell type formation intradural and extramedullary. (c) Represents an extradural component of C2C3 lesion. (d) Anterolateral lesion of smaller size located anterolateral right.
Surgical intervention
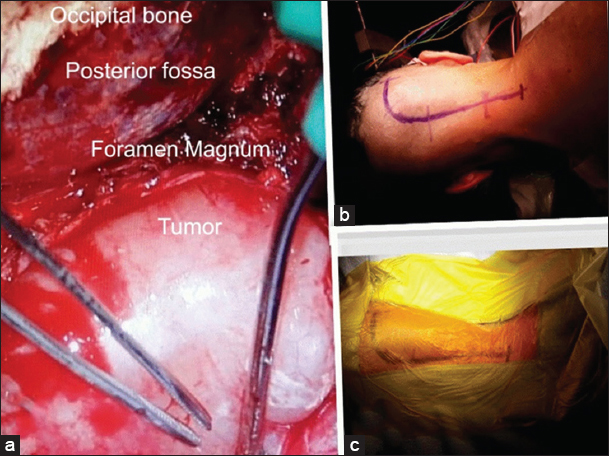
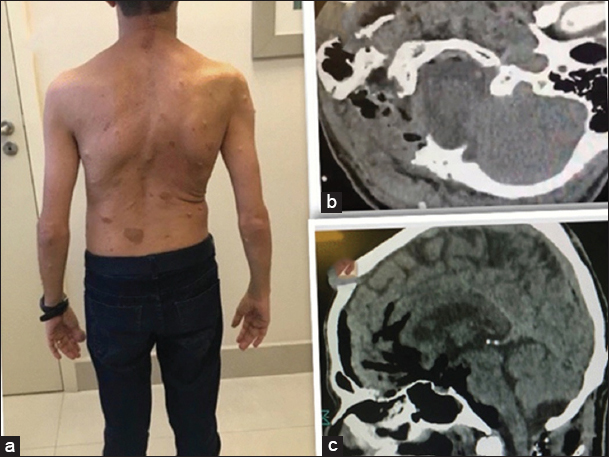
The surgical approach included a right lateral suboccipital craniectomy, removal of the posterior C1 arch, and C2 laminectomy for resection of the dumbbell C2–C3 lesion. This was accompanied by a right C4–C5 hemilaminectomy for excision of the secondary tumor. Gross total lesion resection of the C2–C3 dumbell neurofibroma was accomplished. Partial removal of the C4–C5 secondary lesion was performed as this was benign, and it allowed for the preservation of stability [Figure 3 ]. Somatosensory evoked potential and motor evoked potential monitoring did not change during the procedure. Postoperatively, the patient’s tetraparesis progressively improved and hypoesthesia resolved. The postoperative magnetic resonance (MR) scan obtained revealed satisfactory resection of both lesions [ Figure 4 ].
The immunohistochemistry documented an NF1, for example, negative epithelial membrane antigen (EMA), CD34 positive, KI-67 (MIB-1) negative, positive S100 Protein, and positive neurofilament.
DISCUSSION
Genetic transmission or spontaneous mutation
NF1 may either be genetically inherited (e.g., autosomal dominant) or, more frequently, may occur as a new mutation. The NF1 gene encodes a protein called neurofibromin, which is a tumor suppressor. Changes in expression of the NF1 gene contribute to tumorigenesis and the formation of neurofibromas. They develop around the peripheral nerves, often manifesting as encapsulated masses, and are localized in the dermal and/or subcutaneous tissues. Others may present as plexiform neurofibromas involving peripheral nerve roots.[ 4 ]
Varied phenotypes
NF1 tumors present with varied clinical phenotypes characterized by; café-au-lait spots (95% single/multiple skin lesions) and neurofibromas.[ 3 , 6 ] Plexiform neurofibromas occur in 30% of patients who present with multiple neurofibromas, diffusely involving peripheral nerves. Of the paraspinal neurofibromas, 72% are intradural and extramedullary and 14% are only extradural, while 13% are dumbbell lesions.[ 3 ]
Clinical presentation
With NF1, spinal tumors are occur 40% of the time. Notably, only 2% are symptomatic, often correlated with progressive quadriparesis, along with neck pain and urinary incontinence. Here, the patient had MR-documented medullary compression invading the prebulbar cistern attributed to the C2C3 lesion, plus a secondary C4–C5 tumor contributing to right-sided cord compression.
MR findings of neurofibromatosis 1 lesions
NF1 findings on MRI, as in this case, include iso or hyperintensity on T1, hyperintensity on T2, and homogeneous enhancement with gadolinium of T1 images.[ 4 ]
Pathological/histological and immunohistochemistry findings with neurofibromatosis 1 tumors
Microscopically, NF1 lesions are comprised of Schwann cells, fibroblasts, and perineural cells which typically permeate between/separate the axons from the nerves. The immunohistochemistry commonly shows; S100 positive and EMA negative. Here, the pathological and immunohistochemical results confirmed the diagnosis of NF1; positive immunohistochemistry for CD34, S100 protein, and Ki67, EMA negative, and positive neurofilaments in the intervening axons.
Surgical approach and risks
There are multiple surgical options for dealing with NF1 lesions depending on their spinal location and size. George and Lot proposed a classification system for extramedullary foramen magnum tumors.[ 7 ] The anterior lesions were bilateral or unilateral; the lateral lesions were located between midline and dentate ligament and the posterior lesions behind dentate ligament.[ 7 ] Here, one of the biggest challenges with cervical tumors is the relationship with the vertebral artery (VA) that can be displaced and compressed by the tumor. Therefore, preoperative angiography may be warranted. George and Lot noted the lateral approach (anterolateral and posterolateral) provided the best results versus a standard posterior approach due to the better clinical results and complete lesion ressection. The standard posterior approach is most effective when there the tumor is not adherent to the cord.
Purely extradural tumors anterior to the VA necessitate an anterolateral approach, while most cervical dumbbell warrants a posterolateral approach.
Here, we chose to perform a lateral suboccipital craniectomy, which facilitated identification and dissection away from the VA. The posterior C1 arch was removed, and the C2 laminectomy was performed to achieve complete removal of the C2–C3 lesion. This was followed by a right-sided C4–C5 hemilaminectomy for partial resection of the second C4–C5 lesion (e.g., only partial facet removal to preserve stability).
CONCLUSIONS
In a patient with NF1 who presented with dysphagia/dysphonia and tetraparesis, we removed a giant C2–C3 plexiform/dumbbell neurofibroma extending into the foramen magnum/prebulbar cistern along with a secondary right-sided C5 lesion.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understanding that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Espig A, Slomp A, Campagnolo A, Rockenbach DM, da Silva BD, Pomblum J. Neurofibromatose tipo 1:Atualização. Rev Bras Clin Med. 2008. 6: 243-9
2. Lee MJ, Stephenson DA. Recent developments in neurofibromatosis Type 1. Curr Opin Neurol. 2007. 20: 135-41
3. Ritz G, Batti H, Vigeti N. Neurofibroma plexiforme gigante de dorso:Relato de caso. Arq Catarinenses Med. 2009. 38: 67-9
4. Sarica FB, Cekinmez M, Tufan K, Erdoğan B, Sen O, Altinörs MN. A rare case of massive NF1 with invasion of entire spinal axis by neurofibromas:Case report. Turk Neurosurg. 2008. 18: 99-106
5. Trovó-Marqui AB, Goloni-Bertollo EM, Valério NI. High frequencies of plexiform neurofibromas, mental retardation, learning difficulties, and scoliosis in Brazilien patients with neurofibromatosis Type 1. Braz J Med Biol Res. 2005. 38: 1441-7
6. Yu Y, Hu F, Zhang X, Gu Y, Xie T, Ge J. Application of the hemi-semi-laminectomy approach in the microsurgical treatment of C2 schwannomas. J Spinal Disord Tech. 2014. 27: E199-204
7. George B, Lot G. Neurinomas of the first two cervical nerve roots:A series of 42 cases. J Neurosurg. 1995. 82: 917-23
8. Korf BR, Rubenstein AE.editorsNeurofibromatosis:A Handbook for Patients, Families, and Health Care Professionals. New York: Thieme Medical Publisher; 2005. p.
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου