A rare case of an intramedullary metastasis of a myxopapillary ependymoma
Category: Unique Case Observations
Article Type: Case Report
Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos
Department of Neurosurgery, Centro Hospitalar Lisboa Central-Hospital São José, Serrano, Lisboa, Portugal, Barcelona, Spain.
Department of Neurosurgery, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
Department of Pathology, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
Correspondence Address:
Lino Fonseca
Department of Neurosurgery, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
DOI:10.25259/SNI-96-2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
How to cite this article: Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos. A rare case of an intramedullary metastasis of a myxopapillary ependymoma. 10-May-2019;10:83
How to cite this URL: Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos. A rare case of an intramedullary metastasis of a myxopapillary ependymoma. 10-May-2019;10:83. Available from: http://surgicalneurologyint.com/surgicalint-articles/a-rare-case-of-an-intramedullary-metastasis-of-a-myxopapillary-ependymoma/
Category: Unique Case Observations
Article Type: Case Report
Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos
Department of Neurosurgery, Centro Hospitalar Lisboa Central-Hospital São José, Serrano, Lisboa, Portugal, Barcelona, Spain.
Department of Neurosurgery, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
Department of Pathology, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
Correspondence Address:
Lino Fonseca
Department of Neurosurgery, Hospital Universitari Vall d’Hebron, Barcelona, Spain.
DOI:10.25259/SNI-96-2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
How to cite this article: Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos. A rare case of an intramedullary metastasis of a myxopapillary ependymoma. 10-May-2019;10:83
How to cite this URL: Lino Fonseca, Marta Cicuendez, Francisco Martínez-Ricarte, Elena Martínez-Saez, Esteban Cordero, Agustín Bescos. A rare case of an intramedullary metastasis of a myxopapillary ependymoma. 10-May-2019;10:83. Available from: http://surgicalneurologyint.com/surgicalint-articles/a-rare-case-of-an-intramedullary-metastasis-of-a-myxopapillary-ependymoma/
Background: Myxopapillary ependimoma (MPE) is a benign slow-growing tumor, and it has been designated histologically as a Grade I neoplasm according to the 2016 World Health Organization classification. Despite the benign character, dissemination and metastasis have occasionally been reported. The retrograde dissemination to other levels of the neuraxis is extremely rare, being more frequent to the intracranial compartment.
Case Description: We hereby present a case of medullary metastasis of cauda equina MPE, with a history of having undergone a subtotal resection and postoperative adjuvant radiotherapy. The patient presents complaints of night dorsal pain attributable to intradural metastasis twenty-one years after the first surgical intervention.
Conclusion: The case reported highlights the importance of long follow-up in patients with MPE, since the possibility of secondary seeding to distant craniospinal sites or local spinal sites after surgery, and radiotherapy should be considered in metastatic disease.
Keywords: Ependymoma, intramedullary, metastasis, myxopapillary
INTRODUCTION
Myxopapillary ependymoma (MPE) was first described as a distinct subtype of ependymoma by Kernohaw in 1932.[ 11 , 16 ] MPE is characterized as a slow-growing tumor and classified as Grade I according to the World Health Organization (WHO).[15 ] This subtype accounts for 0.5% of all ependymomas, thus having an incidence of 0.01 per one million inhabitants.[ 20 ] It constitutes 13% of all spinal ependymomas and up to 90% of all conus medullaris tumors.[ 4 ] MPE is located almost exclusively in the conus medullaris, cauda equina, or filum terminale region and is rarely found as a multifocal type, with only little potential for dissemination.[ 2 ] No specific risk factors have been identified although associations with neurofibromatosis type 2 have been suggested.[ 20 ] The prognosis of MPE is very good, with overall 1-year and 10-year survival rates of 100% and 93%, respectively.[ 19 ] The treatment of choice is total surgical excision; however, local recurrence can occur in up to 15% of the cases being the major cause of therapeutic failure. The retrograde dissemination to other levels of the neuraxis is extremely rare, being described more frequently to the intracranial compartment.[ 13 ] We report a case of medullary metastasis of cauda equina MPE and present a brief review of the literature.
CASE REPORT
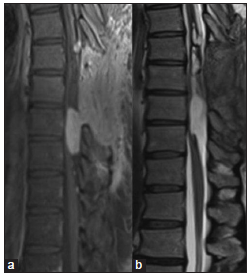
A 43-year-old Caucasian male started experiencing progressive dorsalgia, including nocturnal pain, and progressive resistance to analgesic therapy. He had previoulsy undergone lumbar spine surgery 21 years ago for MPE of cauda equina. A subtotal resection of the tumor was done and postoperative radiotherapy was performed with follow-up scans showing no evidence of residual lesion or any new lesion in the spinal cord. He reported no motor, numbness, bowel, or bladder dysfunction. The patient’s actual neurological examination was normal with no pyramidal or cauda equina compression signs. Magnetic resonance (MR) imaging studies showed an intramedullary lesion with expansive features extending from D5 to D10. The lesion had a cranial cystic and a caudal solid component. After the injection of gadolinium, there was an intense homogenous enhancement of the solid component and several small pachymeningeal lesions throughout the dorsal region and filum terminale. MR studies of the brain and cervical spine showed no abnormality [ Figures 1 and 2 ]. The patient underwent surgical resection under intraoperative neurophysiologic monitoring, including somatosensory evoked potentials and motor evoked potentials. Bilateral D8 and D9 laminectomies were done, and a large, vascularized tumor was encountered intramedullary. We performed a subtotal resection of the solid component because the patient presented a decrease of motor evoked potentials during tumor resection [ Figure 3 ].
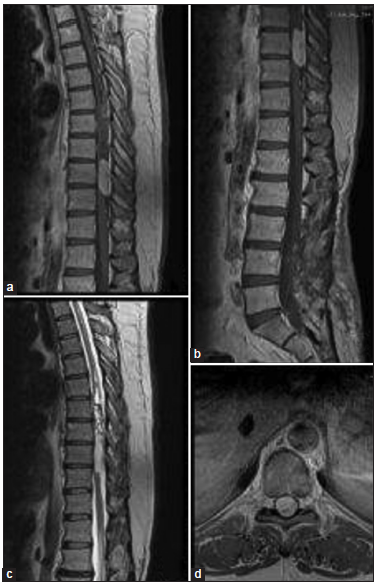
Figure 1
Preoperative magnetic resonance images (MRI). (a) Sagittal T1 sequence with gadolinium contrast showing an intramedullary lesion at D9–D10 level with homogeneous enhacement and several small dorsal implants. (b) Sagittal T1 sequence with contrast where there is no evidence of residual lesion in filum terminale of the first surgery. (c) Sagittal T2 sequence showing an intramedullary isointense lesion in D9–D10 level with hemosiderin focus. (d) Axial T1 Axial MRI T1 with gadolinium contrast at D9 level where the lesion involves the whole spinal cord.
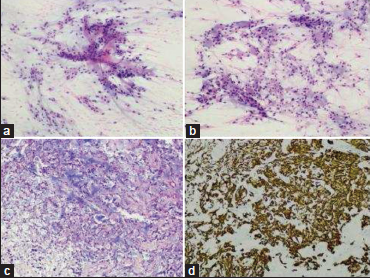
Figure 2
Smears of the specimen showed a papillary pattern with myxoid background (a, H and E, ×200) and presence of globules of myxoid substance (b, H and E, ×200). The paraffin sections reflexed these findings, with a microcystic myxoid background (c, H and E, ×200) and intense glial fibrillary acidic protein (GFAP) expression (d, GFAP, ×200).
Histologically, the lesion was composed of cells with medium- sized, oval nuclei with finely granular chromatin and occasional intranuclear pseudoinclusions. Also well defined eosinophilic cytoplasm with microcystic inclusions and basophilic myxoid material under Alcian-Blue staining. There were frequent hyalinized vessels. No mitotic activity, necrosis, or vascular proliferation was observed. Neoplastic cells expressed glial fibrillary acidic protein and were negative for cytokeratin AE1/ AE3 and EMA. Ki-67 proliferation index was inferior to 2%. A diagnosis of MPE was made (2016 WHO grade I).
The patient did not experience any new postoperative neurological deficits after surgery and he had an uneventful postoperative course. Adjuvant radiotherapy was planned after surgery.
DISCUSSION
Ependymal tumors originate from ependymal cell rests and are uncommon central nervous system neoplasms with an incidence of 0.2/100,000 person-years and a slight predominance in men and Caucasians.[ 5 , 16 ] It has been described a slight upward trend over the past 35 years.[ 5 ] Ependymal tumors usually appear in the fourth decade of life, and in adults, 75% occur in the spinal canal, making up 25% of intramedullary spinal cord tumors and 2% of primary central nervous system neoplasms.[ 3 , 6 ] No specific risk factors have been identified; however, associations with neurofibromatosis type 2, SV-40 polyomavirus exposure, and the lack of vitamin intake in the prenatal period by the mother have been suggested.[20 ]
Ependymomas are divided into three histological entities according to the 2016 WHO classification: ependymoma, subependymoma, and MPE.[ 14 ] Regarding their degree of malignancy and their histological appearance, the WHO defined three grades: MPE and subependymoma are Grade I lesions, the most benign in histologic appearance; Grade II ependymoma includes classic, cellular, papillary, clear cell, and tanycytic subtypes, grouped together for their similar biologic behavior and Grade III are anaplastic ependymomas, which the most malignant behavior.[ 14 ] The grades differ between them in terms of tendency of recurrence, ease of surgical resection, and most usual locations within the spinal cord.[ 6 ] However, recent studies demonstrated that supratentorial location, younger age, genetic markers, and failure to achieve gross total resection may be more accurate predictors for prognosis than histologic grade.[ 17 , 18 , 22 ]
As already mentioned, 75% of ependymomas in the adult population occur in the spinal cord, and 50% of these lesions are MPE.[ 15 ] The conus medullaris and cauda equina region were identified as exclusive sites of MPE.[ 14 ] In the reported case, the tumor was initially located at the cauda equina but then presented as a multifocal type at dorsal segment at time of progression. As a Grade I tumor, MPE is considered to be a slow-growing lesion with benign behavior. Despite these facts, recurrence after both subtotal and gross total resection is well documented.[ 9 , 12 ] Sonneland et al. reported a recurrence rate of 10% for completely resected tumors and 19% for subtotally removed or fragmented tumors, in a population of 77 cases with MPE.[ 21 ] Metastatic behavior has also been reported in the literature, with the intracranial compartment being the most frequent spread site. Metastases have been reported up to 20 years after initial treatment,[ 10 ] emphasizing the role for long-term follow-up in patients treated for MPE. Previously reports of drop metastases in pediatric and adult patients with MPE were reported,[ 8 , 9 ] being a well-established way of dissemination. A recent review by Chakraborti et al.[ 7 ] in 2012 described 19 cases with a variety of intracranial spread sites including the telencephalon, brainstem, and cerebellum, showing the tendency of MPE to spread rostrally in the central nervous system. Although dissemination to the intracranial compartment is well known, the same phenomenon is not established for spinal metastasis. To our knowledge, there are only three reports of secondary spinal dissemination, a single pediatric case, and two adults, described by Libório et al. in 2001.[ 13 ] These appear to be the only reports documenting spinal metastasis of an MPE. Our case is an additional report supporting the possibility of this extremely rare phenomenon.
Despite the benign histology and slow-growing nature of most MPE tumors, some behave in an aggressive manner, with local recurrence, primary seeding, and metastasis. In the management of these patients, radiation therapy and chemotherapy are a treatment option after surgery.[ 22 ] The gold standard first-line therapy for all spinal ependymomas is gross total resection, but when this resection is not possible, adjunctive radiotherapy is recommended.[ 6 ] Sonneland et al. found in an adult series that radiotherapy improves outcome in cases of subtotal resection or metastases.[ 21 ] However, this treatment efficacy is not clear in cases of gross total resection.[ 21 ] Fassett et al., in 2005, describe progression of disease with local recurrence of metastatic behavior in patients treated with radiotherapy.[ 9 ] The explanation for this event is probably due to the localized treatment that would not prevent the spread of the disease, like in our case. Craniospinal irradiation can be considered for patients with disseminated or metastatic disease despite studies are few in number and report mixed results.[ 6 ] The role of chemotherapy is even clearer than radiation, and some studies suggest this treatment as salvage therapy for recurrence if both surgery and radiotherapy fail.[ 6 ] Histologically, MPE metastasis does not show features indicative of the malignancy,[ 1 ] and Ki-67 labeling indices have always been low,[ 7 ] like our case. Therefore, they cannot be used as predictive markers for disease progression.
CONCLUSION
Neurosurgeons should be aware of the possibility of secondary seeding of an MPE to distant craniospinal sites or local spinal sites after surgery and should consider craniospinal imaging as part of preoperative workup and postoperative follow-up. Our case highlights the importance of long follow-up in patients with MPE. Although the limited data, radiotherapy should be considered in patients with metastatic disease.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
References
1. Al-Hussaini M, Herron B. Metastasizing myxopapillary ependymoma. Histopathology. 2005. 46: 469-70
2. Andoh H, Kawaguchi Y, Seki S, Asanuma Y, Fukuoka J, Ishizawa S. Multi-focal myxopapillary ependymoma in the lumbar and sacral regions requiring cranio-spinal radiation therapy: A case report. Asian Spine J. 2011. 5: 68-72
3. Arnautovic K, Arnautovic A. Extramedullary intradural spinal tumors: A review of modern diagnostic and treatment options and a report of a series. Bosn J Basic Med Sci. 2009. 9: 40-5
4. Bagley CA, Wilson S, Kothbauer KF, Bookland MJ, Epstein F, Jallo GI. Long term outcomes following surgical resection of myxopapillary ependymomas. Neurosurg Rev. 2009. 32: 321-34
5. Bates JE, Peterson CR, Yeaney GA, Walter KA, Lundquist T, Rosenzweig D. Spinal drop metastasis in myxopapillary ependymoma: A case report and a review of treatment options. Rare Tumors. 2014. 6: 5404-
6. Celano E, Salehani A, Malcolm JG, Reinertsen E, Hadjipanayis CG. Spinal cord ependymoma: A review of the literature and case series of ten patients. J Neurooncol. 2016. 128: 377-86
7. Chakraborti S, Govindan A, Alapatt JP, Radhakrishnan M, Santosh V. Primary myxopapillary ependymoma of the fourth ventricle with cartilaginous metaplasia: A case report and review of the literature. Brain Tumor Pathol. 2012. 29: 25-30
8. De Falco R, Scarano E, Di Celmo D, Civetta F, Guarnieri L. Concomitant localization of a myxopapillary ependymoma at the middle thoracic part of the spinal cord and at the distal part of the filum terminale. Case report. J Neurosurg Sci. 2008. 52: 87-91
9. Fassett DR, Pingree J, Kestle JR. The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg. 2005. 102: 59-64
10. Fegerl G, Marosi C. Stabilization of metastatic myxopapillary ependymoma with sorafenib. Rare Tumors. 2012. 4: e42-
11. Kernohaw JW.editors. Primary tumors of the spinal cord and intradural filum terminale. In: Penfield W. editor. Cytology and Cellular Pathology of the Nervous System. New York: Paul B Hoeber; 1932. Vol. 3: 993-1025
12. Khan NR, VanLandingham M, O’Brien T, Boop FA, Arnautović K. Primary seeding of myxopapillary ependymoma: Different disease in adult population? Case report and review of literature. World Neurosurg. 2017. 99: 812.e21-8
13. Libório R, Pais RF, Soares GB, Rocha A, Ferreira F, Garcia T. Medullary and intracranial metastases of myxopapillary ependymoma. Acta Med Port. 2001. 14: 133-8
14. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007. 114: 97-109
15. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
16. McGuire CS, Sainani KL, Fisher PG. Incidence patterns for ependymoma: A surveillance, epidemiology, and end results study. J Neurosurg. 2009. 110: 725-9
17. Oh MC, Kim JM, Kaur G, Safaee M, Sun MZ, Singh A. Prognosis by tumor location in adults with spinal ependymomas. J Neurosurg Spine. 2013. 18: 226-35
18. Oh MC, Tarapore PE, Kim JM, Sun MZ, Safaee M, Kaur G. Spinal ependymomas: Benefits of extent of resection for different histological grades. J Clin Neurosci. 2013. 20: 1390-7
19. Reni M, Gatta G, Mazza E, Vecht C. Ependymoma. Crit Rev Oncol Hematol. 2007. 63: 81-9
20. Rege SV, Narayan S, Patil H, Songara A. Spinal myxopapillary ependymoma with interval drop metastasis presenting as cauda equina syndrome: Case report and review of literature. J Spine Surg. 2016. 2: 216-21
21. Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985. 56: 883-93
22. Yang I, Nagasawa DT, Kim W, Spasic M, Trang A, Lu DC. Chromosomal anomalies and prognostic markers for intracranial and spinal ependymomas. J Clin Neurosci. 2012. 19: 779-85
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου